Pancreatic neuroendocrine tumors, also called pNETs or PanNETs, are the second most common type of pancreatic tumor, representing up to 2% of all pancreatic cancer cases. pNETs are distinct from the most common type of pancreatic cancer (pancreatic ductal adenocarcinoma or PDAC) as they tend to be slower-growing, and have a generally better prognosis. Due, in part, to technical advances as well as more frequent imaging, the number of people being diagnosed with pNETs has been increasing. Here we will address what pNETs are, who is at risk for developing them, and how they are classified. In part 2, we will talk about the types of treatments commonly offered to patients as well as emerging techniques and areas where translational research is focusing.
According to the National Cancer Institute’s Surveillance, Epidemiology, and End Result (SEER) Program
4,300–4,400
1.3 / 100,000
60.9 years
Five-Year Survival Rates
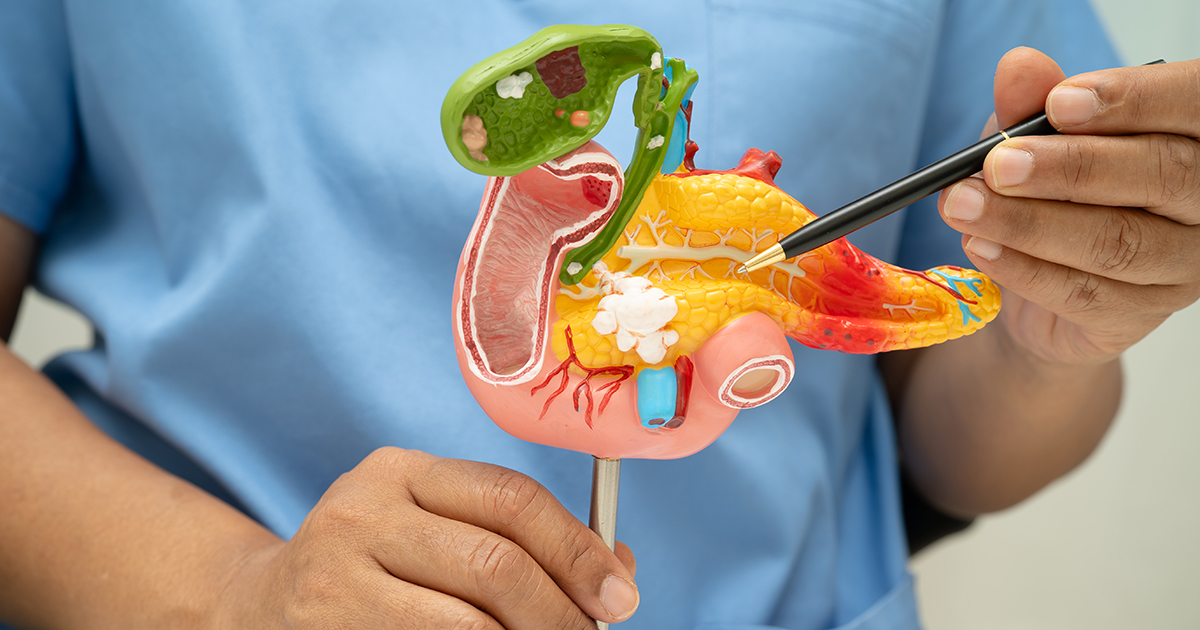
What Are Neuroendocrine Tumors?
Neuroendocrine cells are specialized cells found in many organs. They function by receiving signals from the nervous system and respond by releasing hormones. When these cells acquire the capacity for uncontrolled cell growth, they give rise to neuroendocrine tumors (NETs). NETs are classified based on their site of origin, with the most common locations being the lungs and the gastroenteropancreatic (GEP) system. Within the GEP organs, the pancreas is one of the most frequent sites, accounting for approximately 7% of all neuroendocrine tumors. Neuroendocrine tumors can also arise in other organs, including the thyroid, skin, adrenal glands, and the pituitary gland.
What Are the Different Types of pNETs?
Neuroendocrine tumors are described as either functional or non-functional based on whether they secrete excess hormones.
Non-Functional pNETs
Non-functional pNETs are the most common subtype and are estimated to account for approximately 50–90% of all pNET cases. Unlike functional tumors, they do not secrete excess hormones and therefore typically produce fewer early clinical symptoms. When symptoms do occur — such as weight loss, jaundice, and abdominal pain — they often resemble those seen in PDAC and may go unnoticed or only become evident as the tumor grows or spreads. As a result, non-functional pNETs are frequently diagnosed at more advanced stages compared with functional pNETs.
Functional pNETs
Functional pNETs produce excess hormones and are classified into subtypes according to the specific hormone that they secrete. Because of this abnormal hormone production, functional pNETs often cause more pronounced symptoms or hormone-related syndromes, which can facilitate earlier detection and diagnosis. The most common types of functional pNETs are:
| Type of pNET | Frequency* | Hormone(s) Secreted | Associated Conditions / Symptoms |
|---|---|---|---|
| Insulinoma | 50–60% | Insulin | Low blood sugar and hypoglycemia |
| Gastrinoma | 20–30% | Gastrin | Zollinger-Ellison Syndrome: ulcers and diarrhea |
| Glucagonoma | 5–10% | Glucagon | Severe rash (necrolytic migratory erythema), diabetes, weight loss, and anemia |
| VIPoma | 3–5% | Vasoactive intestinal peptide (VIP) | WDHA syndrome: watery diarrhea, low potassium, and reduced gastric acid |
| Somatostatinoma | 5% | Somatostatin | Somatostatinoma syndrome: weight loss, pain, diabetes, gallstones, diarrhea, and fatty stool |
*Frequency of functional pNETs. pNETs overproducing other hormones are rare and make up ~5% of functional pNETs.
Is There a High-Risk Group for pNET Development?
Most pNETs arise sporadically in people with no family history of neuroendocrine tumors. However, ~10% of pNET patients carry inherited genetic mutations that increase their risk of developing pNETs and other cancers. The two hereditary syndromes most strongly associated with pNET development are:
- Multiple Endocrine Neoplasia type 1 (MEN1) — caused by mutations in the MEN1 gene. Approximately 30–80% of individuals will develop a pNET, most commonly a non-functioning tumor or an insulinoma.
- Von Hippel-Lindau (VHL) Syndrome — caused by mutations in the VHL gene. Between 11–17% of patients develop pNETs, which are typically non-functioning, slow-growing, and have a relatively low risk of spreading.
Less commonly, pNETs can also occur in individuals with Tuberous Sclerosis (TSC), Multiple Endocrine Neoplasia type 4 (MEN4), and Neurofibromatosis type 1 (NF1).
How Are pNETs Classified?
Neuroendocrine tumors are typically classified based on how much cell division is taking place within the tumor. At the time of diagnosis, more than 90% of pNETs are categorized as G1 (low grade) or G2 (intermediate grade), with over 60% of tumors classified as G1. Less than 10% of tumors are designated G3 (high grade).
G3 tumors are further classified according to how closely the tumor cells resemble normal neuroendocrine cells. Well-differentiated tumors, whose cells resemble typical neuroendocrine cells, are called neuroendocrine tumors (NETs). In contrast, tumors with cells that appear atypical or poorly resemble neuroendocrine cells are considered “poorly differentiated” and are classified as neuroendocrine carcinomas (NECs). G3 pancreatic NECs (PanNECs) are generally more aggressive than pNETs and have a worse prognosis.
Spread at Diagnosis
Like other solid tumors, pNETs can be described based on whether they have spread from the pancreas:
~60% Localized — tumor is confined to the pancreas
~25% Regional — spread to nearby lymph nodes or surrounding tissues
~20% Distant — metastatic spread to distant organs

